Carl Kingsford, Carnegie Mellon University, “Transcriptomic Structural Variation Detection”
Jump to:
Bio
“Transcriptomic Structural Variation Detection”

Dr. Carl Kingsford is a Professor in the Computational Biology Department in the School of Computer Science at Carnegie Mellon University (CMU). He received his Ph.D. in Computer Science from Princeton University in 2005 and was an assistant professor at the University of Maryland, College Park, before joining CMU. His main research interests involve machine learning and algorithm design to solve key data analysis problems in computational systems biology and genomics. He and his group have developed new algorithmic techniques for quantifying gene expression, detecting genomic mutations, predicting protein function, reconstructing ancient biological pathways, improving genome assembly, among other problems. He has co-authored 80+ scientific publications. He and his collaborators have developed several widely-used bioinformatics analysis software packages, especially in the area of gene expression.
![]() Click here to view webcast.
Click here to view webcast.
Abstract
“Transcriptomic Structural Variation Detection”
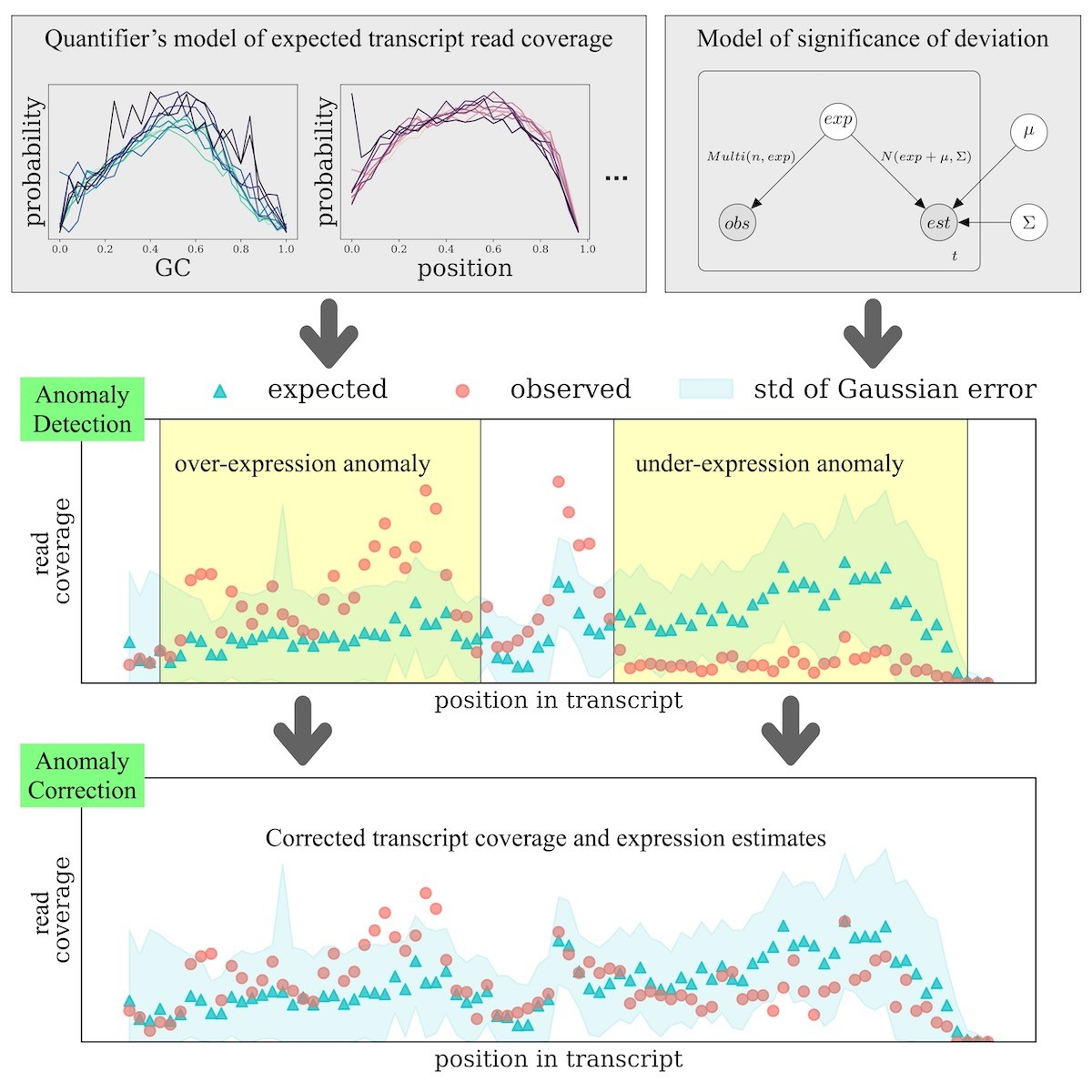
Genomic structural variants that affect expressed transcripts have been implicated in cancer and other diseases. However, detecting these transcriptomic structural variants is challenging computationally. I will describe several computational methods for identifying structural variants from short-read RNA sequencing data. These methods come in two variations. The first type identifies a set of genomic rearrangements that best explains the observed reads and does so in an annotation agnostic way. This allows for the detection of both fusion genes and rearrangements that are not fusions genes but that nevertheless change the sequence of a transcript. The second type of method I will describe identifies coverage anomalies, defined as regions of transcripts where the observed read coverage differs significantly from what one would expect given a sequencing bias model. This is joint work with Cong Ma, Yutong Qiu, and Mingfu Shao.
![]() Click here to view webcast.
Click here to view webcast.