Smita Krishnaswamy, Yale University, “Manifold Learning Yields Insight into Cellular State Space under Complex Experimental Conditions”
Jump to:
Bio
“Manifold Learning Yields Insight into Cellular State Space under Complex Experimental Conditions”

Dr. Smita Krishnaswamy is an Assistant Professor in the department of Genetics at the Yale School of Medicine and Department of Computer Science in the Yale School of Applied Science and Engineering. She is also affiliated with the Yale Center for Biomedical Data Science, Yale Cancer Center, and Program in Applied Mathematics. Smita’s research focuses on developing unsupervised machine learning methods (especially graph signal processing and deep-learning) to denoise, impute, visualize and extract structure, patterns and relationships from big, high throughput, high dimensional biomedical data. Her methods have been applied variety of datasets from many systems including embryoid body differentiation, zebrafish development, the epithelial-to-mesenchymal transition in breast cancer, lung cancer immunotherapy, infectious disease data, gut microbiome data and patient data.
Smita teaches three courses: Machine Learning for Biology (Fall), Deep Learning Theory and applications (spring), Advanced Topics in Machine Learning & Data Mining (Spring). She completed her postdoctoral training at Columbia University in the systems biology department where she focused on learning computational models of cellular signaling from single-cell mass cytometry data. She was trained as a computer scientist with a Ph.D. from the University of Michigan’s EECS department where her research focused on algorithms for automated synthesis and probabilistic verification of nanoscale logic circuits. Following her time in Michigan, Smita spent 2 years at IBM’s TJ Watson Research Center as a researcher in the systems division where she worked on automated bug finding and error correction in logic.
![]() Click here to view webcast.
Click here to view webcast.
Abstract
“Manifold Learning Yields Insight into Cellular State Space under Complex Experimental Conditions”
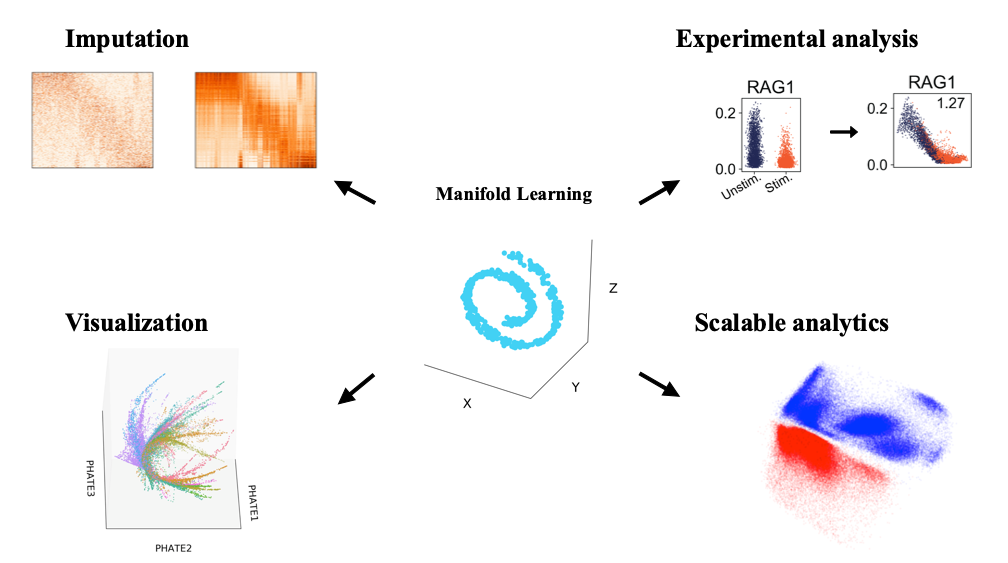
Recent advances in single-cell technologies enable deep insights into cellular development, gene regulation, cell fate and phenotypic diversity. While these technologies hold great potential for improving our understanding of cellular state space, they also pose new challenges in terms of scale, complexity, noise, measurement artifact which require advanced mathematical and algorithmic tools to extract underlying biological signals. Further as experimental designs become more complex, there are multiple samples (patients) or conditions under which single-cell RNA sequencing datasets are generated and must be batch corrected and the corresponding populations of single cells compared. In this talk, I cover one of most promising techniques to tackle these problems: manifold learning. Manifold learning provides a powerful structure for algorithmic approaches to denoise the data, visualize the data and understand progressions, clusters and other regulatory patterns, as well as correct for batch effects to unify data. I will cover two alternative approaches to manifold learning, graph signal processing (GSP) and deep learning (DL), and show results in several projects including: 1) MAGIC (Markov Affinity-based Graph Imputation of Cells): an algorithm that low-pass filters data after learning a data graph, for denoising and transcript recover of single cells, validated on HMLE breast cancer cells undergoing an epithelial-to-mesenchymal transition. 2) PHATE (Potential of Heat-diffusion Affinity-based Transition Embedding): a visualization technique that offers an alternative to tSNE in that it preserves local and global structures, clusters as well as progressions using an information-theoretic distance between diffusion probabilities. 3) MELD (Manifold-enhancement of latent variables): an analysis technique that filters the experimental label on the graph learned from single-cell data in order to boost experimental signal and associated correlations. 4) SAUCIE (Sparse AutoEncoders for Clustering Imputation and Embedding), our highly scalable neural network architecture that simultaneously performs denoising, batch normalization, clustering and visualization via custom regularizations on different hidden layers. We demonstrate the power of SAUCIE on a massive single-cell dataset consisting of 180 samples of PBMCs from Dengue patients, with a total of 20 million cells. We find that SAUCIE performs all the above tasks efficiently and can further be used for stratifying patients themselves on the basis of their single cell populations. Finally, I will preview ongoing work in neural network architectures for predicting dynamics and other biological tasks.
![]() Click here to view webcast.
Click here to view webcast.